High-Throughput Virtual Screening and Docking for The Discovery of Antiviral Agents
Keywords:
- Antiviral agents, high-throughput virtual screening, molecular docking, main protease, AutoDock Vina, drug-likeness, ADMET, structure-based drug design.
Abstract
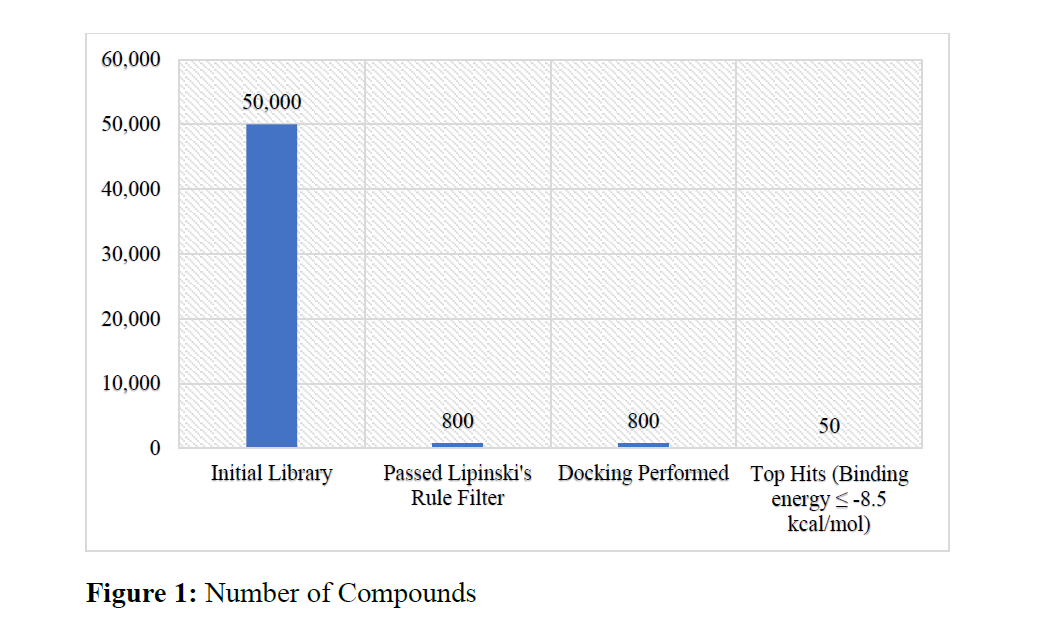
The emergence of viral infections demands the quick identification and development of antiviral producers in a timely and cost-efficient manner. In this study, we implemented a high-throughput virtual screening (HTVS, followed by molecular docking) to test for potential antiviral compounds that could act on the main protease (Mpro) of a model virus. Following drug-likeness criteria using Lipinski’s Rule of Five, we computationally screened through 50,000 small molecules and narrowed our search to 800 drug-like candidates. We then used AutoDock Vina to dock the drug candidates into the Mpro. The docking of the finalists produced 50 compounds with binding energies ≤ -8.5 kcal/mol. Ultimately, compound C_47821 was the most promising lead with the highest binding energy (-10.2 kcal/mol) and the most hydrogen bonds with the active site of Mpro. Analysis with ANOVA confirmed that hydrogen bonding played a significant role in the binding energy differences p < 0.001. ADMET profiling further supported the pharmacokinetic relevance of the top hits. Our results demonstrate how computational pipelines can rapidly expedite early-stage antiviral drug discovery, and we identified lead compounds that would be promising for experimental verification in the lab.