In Silico Molecular Docking Approaches in The Identification of Novel Enzyme Inhibitors
Keywords:
- Molecular docking, enzyme inhibitors, in silico drug discovery, AutoDock Vina, binding affinity, virtual screening, ligand clustering, RMSD validation.
Abstract
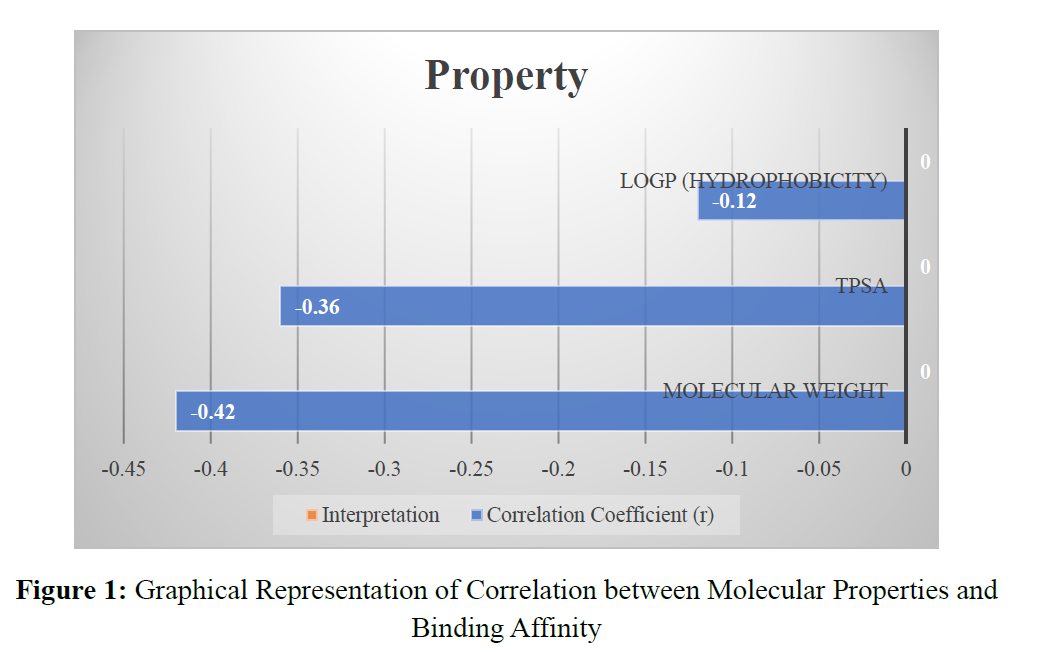
The increasing demand for efficient and targeted therapeutic agents has led to the widespread adoption of in silico molecular docking techniques in modern drug discovery. This study explores the effectiveness of computational docking in identifying novel enzyme inhibitors with potential pharmacological significance. A total of 200 structurally diverse ligands, sourced from ZINC, PubChem, and ChEMBL databases, were screened against a disease-related enzyme target using AutoDock Vina. The binding affinities ranged from -11.3 to -5.4 kcal/mol, with a mean of -7.9 kcal/mol, indicating moderate to strong interactions in the majority of compounds. Cluster and correlation analyses revealed significant relationships between molecular properties (such as molecular weight and TPSA) and docking performance, highlighting key physicochemical contributors to binding affinity. The top 10 ligands demonstrated high-affinity interactions and retained consistent binding poses during redocking validation, confirming the reliability of the docking protocol. Visualization of key interactions, along with RMSD values below 2.0 Å, further validated binding stability. This study underscores the value of molecular docking as a cost-effective and high-throughput strategy in the early stages of drug development, while also acknowledging limitations and suggesting future integration with molecular dynamics and ADMET profiling to enhance predictive accuracy and clinical relevance.