Structure-Based Drug Discovery Using Molecular Dynamics and Docking Techniques
Keywords:
- Structure-Based Drug Discovery, Molecular Docking, Molecular Dynamics Simulation, Binding Affinity, Protein-Ligand Interaction, Drug Design
Abstract
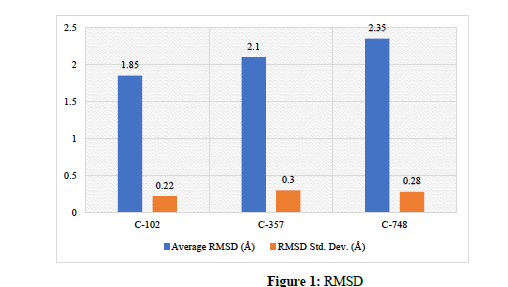
Structure-based drug discovery (SBDD) has emerged as an important means of identifying novel therapeutic candidates using the 3-dimensional structure of target proteins. In this work, molecular docking and molecular dynamics (MD) simulations were applied to 1000 compounds against a target protein aiming to identify potential inhibitors with strong binding affinity and stable binding interactions with the target protein. The top 10 ranked compounds from the docking analysis were further evaluated by applying 100 ns MD simulations to investigate the dynamic stability of the selected compounds according to standard analysis protocols using root mean square deviation (RMSD) and calculation of stable hydrogen bonding interactions. Compound C-102 was identified as the most promising candidate with the lowest RMSD and the greatest average number of hydrogen bonds confirming better stability of the complex with the target protein. Statistical analyses confirmed persistence of significant differences in the stability of the compounds from the selected candidates. Overall study findings support concerns determined using the docking and MD simulation approaches used in this work can fit into the drug discovery process in the early stages and signify a robust, reproducible approach to lead identification and maturation.